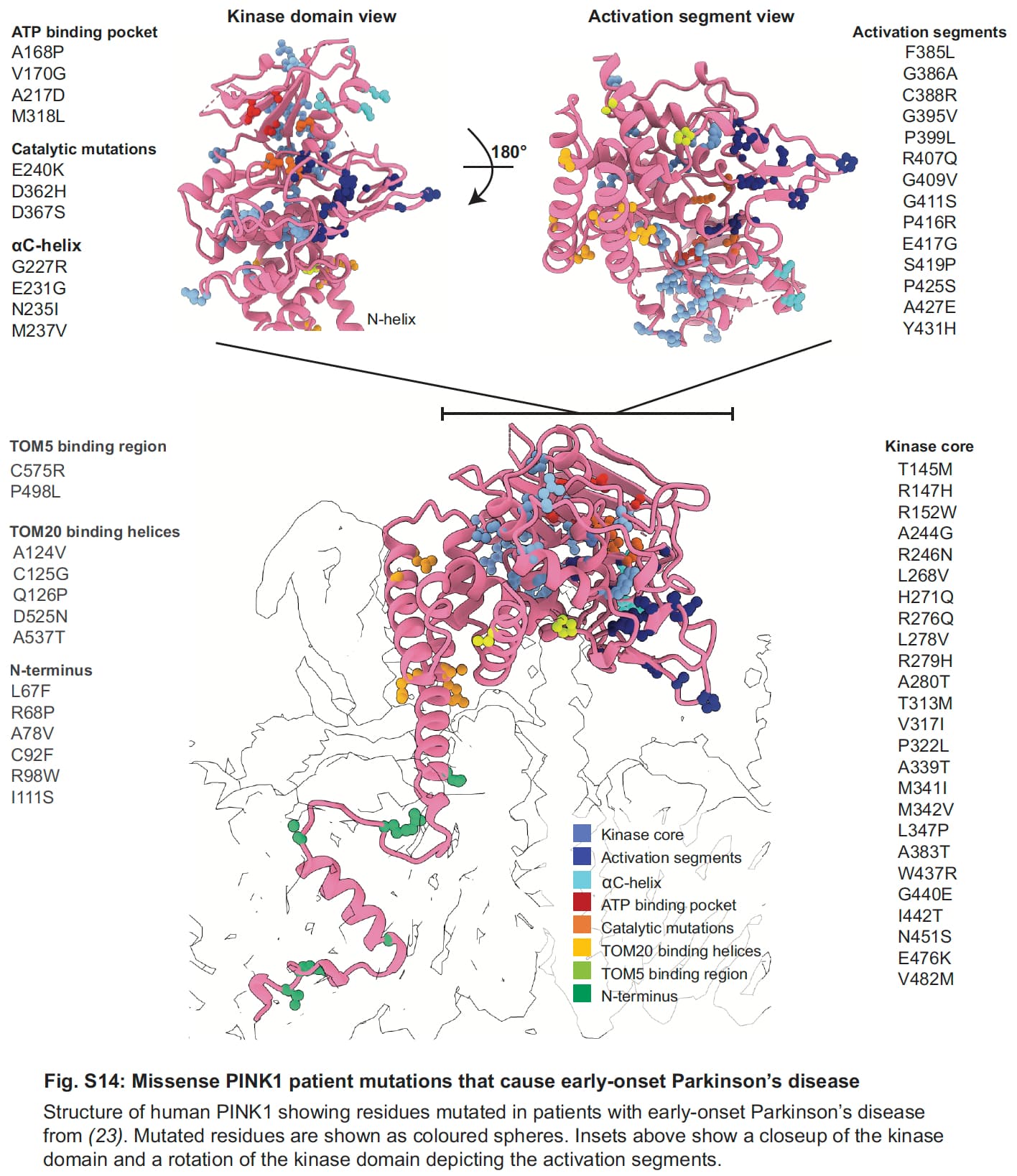
This is huge: Chronobiotic use of melatonin improves DAT-binding in iRBD
Dieter Kunz1, Jan De Zeeuw2, Sophia Stotz1, Michail Plotkin3, Frederik Bes1
1 Charité – Universitätsmedizin Berlin, Institute of Physiology, Berlin, Germany,
2 Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin, Germany,
3 Charité – Universitätsmedizin Berlin, Berlin, Germany
Aims: Isolated REM-sleep behavior disorder (iRBD) is recognized as a prodromal state of clinical α-synucleinopathies such as Lewy-body dementia and Parkinson’s disease. A pathophysiologic hallmark of α-synucleinopathies is nigrostriatal dopaminergic impairment, with dopamine-transporter (DaT)-SPECT imaging considered best available prognostic and monitoring marker. DaT-binding is reported to decrease with healthy aging by 4-10% per decade, accelerated to 4-12% per year iRBD patients. We have introduced melatonin as a treatment option for iRBD. Aim of the study was to evaluate effects of melatonin on DaT-SPECT imaging in iRBD patients.
Methods: In a prospective, longitudinal, observational, single-center study we performed at least two DaT-SPECTs in 97 iRBD patients treated with melatonin as a chronobiotic (i.e. administration always- at-the-the-same-clock-time;10-11p.m.-corrected for chronotype); 28 patients were excluded mainly due to change of psychotropic drugs known to influence DaT.
Results: After mean follow-up of 3.6yrs, only 21/69 patients (11 female; mean age 71±6yrs) showed specific binding ratios (SBR) in most affected region (MAR, predominantly right posterior putamen) comparable to usually reported declines with iRBD. In contrast, 7 had declined SBR at a rate comparable to healthy aging, while 41 had actually improved SBR. Improvement after one year (SBR of MAR; F1,31=23.748;p>0.001) and two years was significant (F1,24=4.648;p=0.041). After four years half of the patients showed a higher SBR than baseline (23 vs. 24 patients), though this was not significant. 47/69 of our patients at baseline met established criteria for an advanced state.
Conclusions: To the best of our knowledge, present data give first evidence for a consistent increase in DaT-binding ratios in nigrostriatum over time in a cohort of patients with iRBD. In addition, the previously reported persisting effect of melatonin on RBD symptoms suggest that melatonin, when used as a chronobiotic, may have a disease-modifying effect in prodromal α-synucleinopathies.
 It’s a conference poster so not peer-reviewed yet but it’s a very serious research team, it’s a decent size, over a long period of time and it uses an objective biomarker
It’s a conference poster so not peer-reviewed yet but it’s a very serious research team, it’s a decent size, over a long period of time and it uses an objective biomarker
Their chronobiotic protocol is “2 mg, ≥6 months, always-at-the-same-clock time, 10-11pm, corrected for chronotype”: Treatment of isolated REM sleep behavior disorder using melatonin as a chronobiotic 2021. In this 2021, paper they note:
Patients on concomitant betablocker or antidepressant therapy seemed to respond more slowly at the beginning of melatonin treatment, although they did not differ in general from the no-confounder group. Both antidepressants and betablockers are known to induce secondary RBD as well as to increase RSWA in patients with iRBD. Antidepressants are well known to influence REM sleep, with anticholinergic agents to suppress, but serotoninergic and noradrenergic agents to spoil the quality of REM sleep. As has been known for decades, lipophilic propranolol blocks melatonin secretion from the pineal gland via beta-receptors. The suppression of melatonin with betablockers predominantly affects REM sleep, which can be reversed by exogenous melatonin. Long-term medication with betablockers is likely to have changed melatonin receptor sensitivity, thus delaying response to initial melatonin. The same negative effect could be attributed to recommended increasing dosage of melatonin. Because melatonin influences its own receptor, it is important to have a melatonin-free period over the day. Supraphysiologic melatonin doses, especially in slow metabolizers, prevent the absence of melatonin during the day and could induce insensitivity in melatonin receptors the next evening.
The rationale for this strict schedule is that since our initial pilot studies with RBD patients about 25 years ago, we repeatedly observed that responders and nonresponders were best distinguished by evaluating their sleep hygiene, that is, stable vs. varying bedtimes and times of melatonin intake (summarized in 10, 17). This clinical observation is in agreement with the fact that melatonin is known to feedback on the suprachiasmatic nucleus, the central pacemaker or master-clock. As a consequence, exogenous melatonin should be administered consistently within a rather narrow time span in order to gain optimal effects. Patients are informed that melatonin in RBD rarely exhibits effects during first days of treatment, rather effects occur within the first weeks. Sometimes symptoms even rapidly worsen over the first days, presumably because appointed time of administration induced a transient initial delay or advance of circadian phase. In those patients, in whom melatonin does not show positive effects over the first 3 weeks of treatment, the time of administration is controlled referring to individual chronotype.
The rate of improvement of RBD symptoms with melatonin in previously reported case series varies, and two recent RCTs have shown no effect. Unfortunately, melatonin has been sold worldwide for the past 25 years as a hypnotic to be administered in connection with clock time independent events (eg, “after a meal,” “at bedtime”). Most people who took melatonin—including those in the two recent RCTs with negative results—will therefore not have adhered to a schedule based strictly on clock time. As an example, in our Clinic for Sleep & Chronomedicine, we precisely explain the chronobiotic protocol but even though, still some patients stuck to the aberrant leaflet prescription. Our study indeed demonstrates that beneficial effects of melatonin can easily be disrupted with improper timing of intake, which may well explain lower response rates reported by other groups. In those patients for whom we had a chance to reinstruct, melatonin improved RBD symptoms. On the other hand, melatonin should not be considered a harmless drug or being without side effects. Inadequate timing of melatonin seems likely not only to fail in improvement, but rather to worsen symptomatology due to desynchronization.
What do you think about this potential risk @John_Hemming?