How to stop the shift of drug discovery from the U.S. to China
The FDA must make it easier to do such work in the U.S.
By Scott Gottlieb
May 6, 2025
Gottlieb, served as commissioner of the Food and Drug Administration from 2017 to 2019.
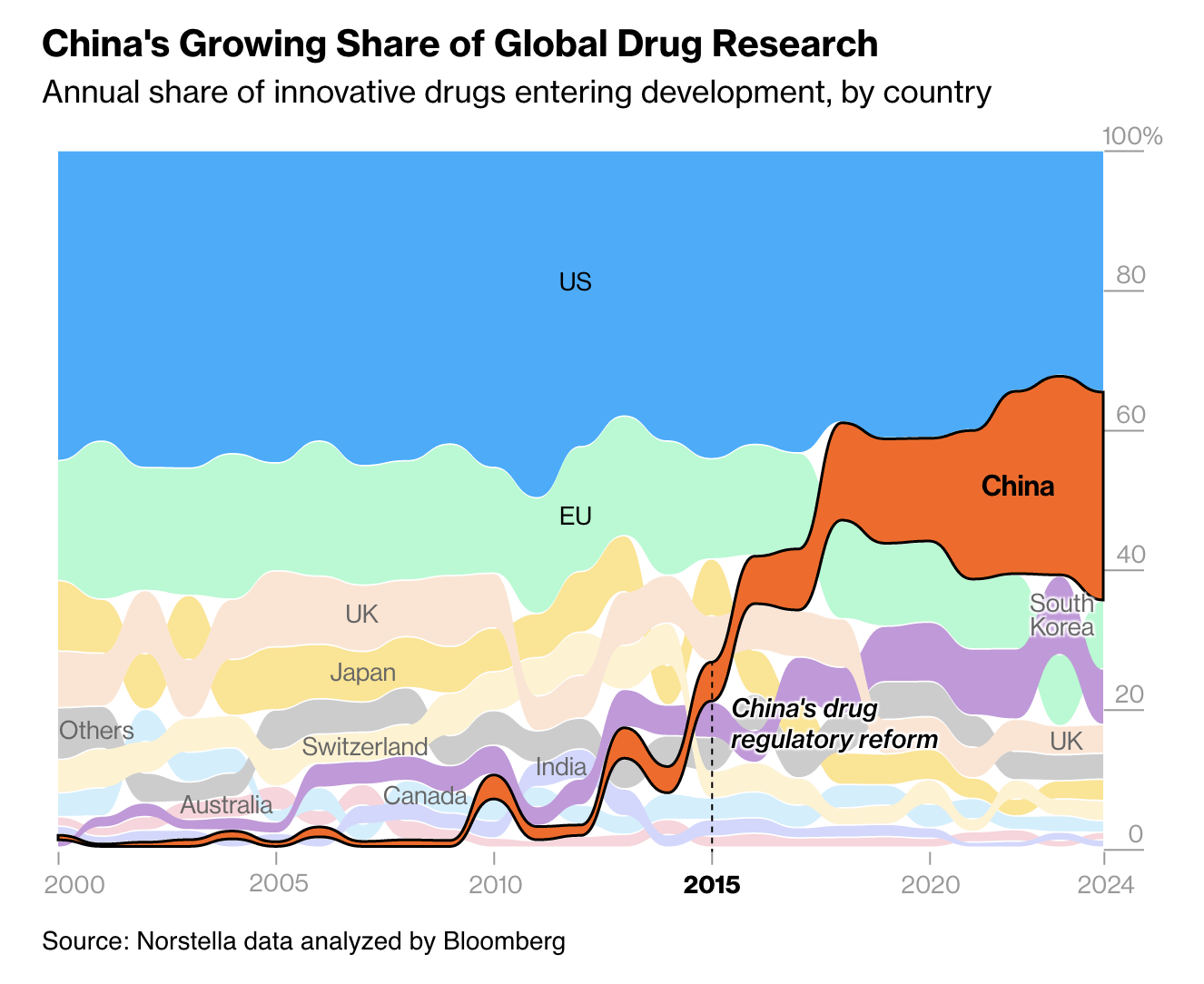
Five years ago, U.S. pharmaceutical companies didn’t license any new drugs from China. By 2024, one-third of their new compounds were coming from Chinese biotechnology firms.
Why are U.S. drugmakers sending their business to China? As in many other industries, it’s so much cheaper to synthesize new compounds inside Chinese biotechnology firms once a novel biological target has been discovered in American laboratories.
Yet the costs of developing new drugs in the U.S. needn’t be so high. They are driven up, in part, by increasing regulatory requirements that burden early-stage drug discovery in America. That’s especially true for Phase I clinical trials, in which drugs are tested in people for the first time.
This shift of discovery work to China is going to accelerate if we don’t take deliberate steps to make it easier to do such work here in America. Yet the imperative to modernize early-stage drug development — to ensure that groundbreaking drug discovery remains in the U.S. rather than migrating to China — is colliding head-on with an impulse to slash the very government workforce capable of spearheading these reforms. These conflicting impulses have created a paradoxical tension: on one hand, the desire to stay competitive with China in biotechnology innovation, and on the other, a parallel campaign to reduce and in some cases dismantle the investments and institutions essential to achieving that goal.
In most cases, Chinese firms are not discovering new biological targets, nor are they crafting genuinely novel compounds to engage these targets through homegrown Chinese research. Instead, they piggyback on Western innovations by scouring U.S. patents, zeroing in on biological targets that are initially uncovered in American labs, and then developing “me too” drugs that replicate American-made compounds with only superficial tweaks, or producing “fast follower” drugs that capitalize on the original breakthroughs while refining key features to try to surpass U.S. innovation. (Ed.: China is copycat!) Facing fewer regulations, the Chinese drugmakers can move more quickly than U.S. biotechnology companies — synthesizing copy-cat drugs based on our biological advances and then promptly moving these Chinese-made compounds into early-stage clinical trials, outpacing their American counterparts.
According to the investment bank Jefferies, large American drug companies spent more than $4.2 billion over the past year licensing or acquiring new compounds originally synthesized by Chinese firms. Many comprised advanced compounds such as [antibody drugs and cell therapies](Is China the future of biotech?) — underscoring Chinese companies’ growing sophistication in adopting the latest American technologies. The cost of licensing these compounds from China, rather than synthesizing them in American labs, can be significantly lower. At a time when research funding in the U.S. is being cut, and research budgets are becoming painfully stretched, companies are looking to lower the cost of building their pipelines. In a fast-moving field such as oncology, this shift toward Chinese-synthesized compounds is particularly striking: I am told by someone inside the FDA process that nearly three-quarters of new small molecule cancer drugs submitted to the Food and Drug Administration for permission to begin U.S.-based clinical trials are initially made in China. (Ed.: China is innovator!)
Usually, only a few months elapse between the moment a U.S. research team publishes a patent identifying a new biological target and when a biotechnology firm in China creates the corresponding drug that capitalizes on these findings. Because Chinese firms can synthesize new molecules at a fraction of the cost incurred by U.S. biotechnology companies — owing to a large and skilled but much cheaper workforce (Ed.: Cheap Chinese labor!) — they find the most intriguing biological targets pursued by Western researchers, rapidly churning out potent yet less expensive copycat molecules that they then market to Western companies.
A major challenge for U.S. firms is the long and costly process of obtaining FDA approval for Phase I studies, in which drugmakers test a new drug’s safety and tolerability in a small group of human volunteers. In China, launching this initial phase of clinical trials is far simpler, giving Chinese biotechnology companies a competitive advantage: By swiftly advancing their molecules into early-stage patient testing, Chinese firms can more readily determine which compounds hit their biological targets and show the greatest therapeutic promise. This allows the Chinese firms to quickly refine their molecules and then leapfrog their American counterparts, who are slowed by more cautious regulatory processes. While China’s regulatory process doesn’t uphold the patient safeguards that Americans rightly insist upon, the U.S. FDA could still streamline its path into early-stage drug development, bolstering America’s competitive edge without compromising patient safety.
In the U.S., one of the costliest early hurdles is the exhaustive animal testing that the FDA requires before a drug can be advanced into Phase I studies. These “pre-clinical” studies help safeguard patients, but the agency also uses this testing to weed out potential failures before a drug requires more intensive FDA scrutiny in later trials.
Over time, this regulatory framework has frontloaded a significant share of costs to the earliest phases of drug development, when biotechnology startups are often running on shoestring budgets, lack clinical data to attract investors, and can least afford delays. One measure of the increasing difficulty in securing the FDA’s permission for Phase I trials is the growing number of U.S. drugmakers who take compounds discovered on American soil and conduct these clinical trials in other Western markets, where they can obtain data more quickly and inexpensively before bringing it back to the FDA. One popular locale is Australia, where costs run about 60% lower than U.S.-based clinical trials, largely because the Australian government offers tax incentives to attract this kind of biomedical investment.
Many animal studies address esoteric questions about a drug’s long-term effects on parameters that may not be relevant to its eventual use — for example, at doses and durations of use that may be far beyond how patients will ultimately use the drug. The FDA’s preclinical testing protocols sometimes require American researchers to administer new compounds to animals at levels up to 500 times higher than any intended dose for patients, aiming for maximum animal exposure before human trials can begin. Where the FDA needs to screen for certain remote risks, many animal studies could be safely deferred until human trials confirm that a drug may benefit patients. At that point, it becomes easier for biotechnology companies to raise capital to fund these pro forma testing efforts.
To modernize the process, the FDA could tap into the wealth of data from existing drugs to establish a more phased approach to these requirements, where the amount of initial animal testing is more closely matched to a drug’s novelty and a better estimation of its perceived risks. It’s a prime opportunity to employ artificial intelligence — mining current data and extrapolating known information to newly discovered molecules. For new molecules that share structural similarities with established drugs, where a robust body of safety information already exists (and the likelihood of uncovering novel risks is judged to be minimal), some animal studies might simply be unnecessary. To establish a graduated approach to the scope of pre-clinical toxicology studies that the FDA requires for new molecules, Congress could revise the agency’s statutory framework, explicitly empowering it to adopt such flexible standards. It would also require targeted investments, enabling the FDA to craft the necessary tools and protocols to implement these refined methodologies.
Mice and even primates are often poor proxies for many of the remote toxicities the FDA is trying to test for, anyway. The agency can also make a more concerted effort to adopt advanced technologies, like pieces of human organs embedded in chips that can be used to test for remote dangers a drug may pose to specific organs like the heart and liver. These tools can reliably screen for risks at a fraction of the time and cost. FDA Commissioner Marty Makary recently announced his intention to pursue a plan that would phase out animal studies in the preclinical evaluation of antibody drugs, shifting instead toward innovative technologies that assess toxicology without relying on live animals. This positive step requires the FDA to invest in new capabilities, and scientific staff that possess expertise in these novel domains.
But right now, that investment seems unlikely. The size and scientific scope of the FDA staff responsible for reviewing early-stage drug development — and evaluating data collected from animal studies — has failed to keep up with the increasing complexity and sheer volume of applications flooding into the agency to launch Phase I clinical trials. Now, the FDA has made deep staffing cuts, prompted by DOGE, that have specifically targeted scientific teams that would lead these essential reforms.
Adding to these woes, morale at the FDA has declined so markedly that many foresee a wave of voluntary resignations among clinical reviewers. By thinning the ranks of experts who tackle novel scientific questions and resolve issues that span across different drug development programs — especially the elimination of the policy office within the FDA’s Office of New Drugs, which adjudicated these kinds of cross-cutting scientific questions — the government has impeded the early dialogue with drug developers that often results in streamlining requirements for Phase I studies. Even more challenging, it weakens the staff’s ability to develop new guidance documents and put better review practices into place — reforms essential for lasting improvements to the preclinical review process.
Instead of strengthening America’s biotechnology ecosystem, such measures risk accelerating the migration of discovery activities to China, undermining innovation at home. When U.S. drugmakers license compounds from China, they divert funds that might otherwise bolster innovation hubs such as Boston’s Kendall Square or North Carolina’s Research Triangle. The U.S. biotechnology industry was the world’s envy, but if we’re not careful, every drug could be made in China.
Scott Gottlieb, M.D., is a senior fellow at the American Enterprise Institute and served as commissioner of the Food and Drug Administration from 2017 to 2019. He is a partner at the venture capital firm New Enterprise Associates and serves on the boards of directors of Pfizer Inc. and Illumina.
Grok’s take:
The editorial “How to Stop the Shift of Drug Discovery from the U.S. to China,” published on May 6, 2025, by STAT News and authored by former FDA Commissioner Scott Gottlieb, argues that the U.S. is losing its edge in early-stage drug discovery to China due to regulatory burdens and cost disparities. Gottlieb highlights that by 2024, one-third of new compounds licensed by U.S. pharmaceutical companies came from Chinese biotech firms, a stark shift from zero five years prior. He attributes this to lower costs and faster clinical trials in China, particularly in Phase I studies, and calls for regulatory reform to keep innovation in the U.S. Let’s break down the piece critically, examining its claims, implications, and gaps.
Gottlieb’s core argument is that U.S. regulatory requirements, especially for Phase I trials, frontload costs and delays, pushing startups to outsource to China. He notes that Chinese firms often produce “me-too” or “fast-follower” drugs by building on U.S. discoveries, exploiting American patents to develop compounds with minor tweaks. This is a valid concern—China’s regulatory environment is indeed less stringent, allowing faster trials. For instance, the editorial mentions that U.S. firms often conduct Phase I trials in places like Australia, where costs are 60% lower due to tax incentives, underscoring how FDA processes can deter domestic innovation. The financial strain on U.S. startups, often operating on tight budgets, is real; preclinical animal testing and FDA approvals can take years and millions of dollars, which Gottlieb argues could be streamlined without sacrificing safety.
However, the piece oversimplifies the dynamics. Gottlieb frames China as merely copying U.S. innovation, but this downplays China’s growing capabilities. A February 2025 STAT News article notes that Chinese firms like DeepSeek are leveraging AI to accelerate drug development at a fraction of U.S. costs—DeepSeek’s large language model was trained for under $6 million, compared to billions for OpenAI’s ChatGPT. This suggests China isn’t just piggybacking but building its own technological edge, which Gottlieb doesn’t fully address. Additionally, a post on X from April 24, 2025, by @cremieuxrecueil highlights China’s trial reforms, noting they now match the U.S. in clinical trial numbers and scale, positioning them to lead in pharma soon. This challenges Gottlieb’s narrative of China as a mere imitator—it’s becoming a competitor in its own right.
The editorial also glosses over systemic U.S. issues beyond regulation. The U.S. biotech sector faces structural challenges like a patent system that, as a March 2025 STAT News piece argues, rewards monopolies over innovation, squeezing small startups. Philanthropy is proposed as a fix, but Gottlieb doesn’t explore this or other market-based solutions, focusing instead on FDA reform. This narrow lens ignores broader economic pressures—like venture capital drying up for early-stage biotechs, a trend noted in industry analyses—or the impact of U.S. policies like Trump’s tariffs, which a February 2025 STAT News article warns could raise drug costs and disrupt supply chains, further deterring domestic innovation.
Gottlieb’s call for regulatory modernization is reasonable but lacks specifics. He suggests streamlining preclinical testing and FDA processes, but what does that look like? The FDA’s January 2025 draft guidance on AI in drug development, which Gottlieb doesn’t mention, shows the agency is already adapting to new tech—over 500 submissions since 2016 involved AI, per STAT News. This suggests the FDA isn’t as stagnant as portrayed, though it may still lag behind China’s pace. Gottlieb also fails to address patient safety concerns in detail. While he claims reforms wouldn’t compromise safety, China’s lax regulations have led to ethical issues, as seen in its drug user surveillance policies (a 2022 Health and Human Rights Journal article notes coercive “grid management” of drug users). Speeding up U.S. trials must balance efficiency with rigor, a tension Gottlieb doesn’t fully grapple with.
The editorial’s most glaring oversight is its dismissal of workforce cuts as a barrier to reform. Gottlieb warns that slashing FDA staff—a trend noted in an April 2025 STAT News piece about layoffs slowing drug reviews—undermines the agency’s ability to modernize. Yet he doesn’t connect this to broader political currents, like the Trump administration’s policies. A December 2024 STAT News article notes Trump’s threats to withdraw from the WHO and impose tariffs on Mexico and Canada over fentanyl, reflecting a punitive approach that could exacerbate biotech’s challenges. These policies might drive more offshoring, not less, as firms seek stability elsewhere. Gottlieb’s failure to critique this political context feels like a missed opportunity, especially given his FDA tenure.
Public sentiment on X aligns with Gottlieb’s concerns but adds nuance. Posts from May 6, 2025, by users like @ScottGottliebMD and @Jasonmmast amplify the editorial, warning of the innovation drain to China. However, earlier posts, like @TheChiefNerd’s January 2025 comment, cite Gottlieb himself noting cost disparities as a key driver, suggesting this issue has been brewing for months. The lack of counterarguments on X—no users push back significantly—might reflect agreement but could also indicate a lack of engagement with alternative views, like the potential for global collaboration to drive innovation rather than competition.
The establishment narrative, which Gottlieb largely supports, frames the U.S. as a victim of unfair competition, needing to double down on domestic reform. But this overlooks the benefits of globalized drug development—cheaper drugs from China could benefit U.S. patients, a point raised in a May 2024 STAT News article warning that severing ties with Chinese firms might disrupt supplies for millions. It also ignores how U.S. policies, like excessive patent protections (criticized in an April 2025 STAT News piece), contribute to high costs and push firms abroad. A more balanced approach might involve international cooperation on standards, rather than a protectionist race to the bottom on regulation.
In summary, Gottlieb’s editorial raises a critical issue: the U.S. is losing ground in drug discovery due to regulatory and cost barriers. His call for reform is well-founded, but the piece lacks depth in addressing China’s innovation capacity, systemic U.S. issues, and the political forces shaping biotech. It also doesn’t grapple with the trade-offs of speeding up trials or the potential upsides of global collaboration. The argument is compelling but feels like a starting point, not a comprehensive solution. More data on successful regulatory models—like the European Medicines Agency’s AI reflection paper (September 2024)—and a harder look at U.S. policy failures would strengthen the case.