Re @rapadmin posted Inducing Autophagy by Rapamycin Before, but Not After, the Formation of Plaques and Tangles Ameliorates Cognitive Deficits (2011)
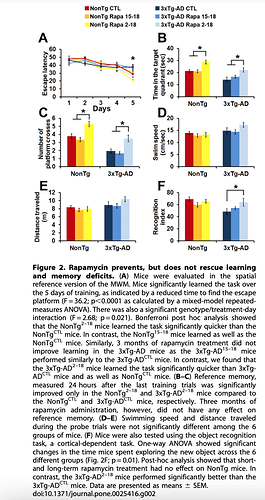
Good fundamental paper looking at a mouse model of amyloid and Rapamycin intervention, including control mice. Yes, it’s one of these transgenic mutant mice models, since mice don’t get amyloid like humans. Rapamycin administration, earlier in life ameliorates cognitive dysfunction of aging, in both wild control and transgenic models. Rapamycin DOES cross the BBB, impacts mTOR, and reduces cognitive decline. "Rapamycin prevents, but does not rescue learning and memory deficits"
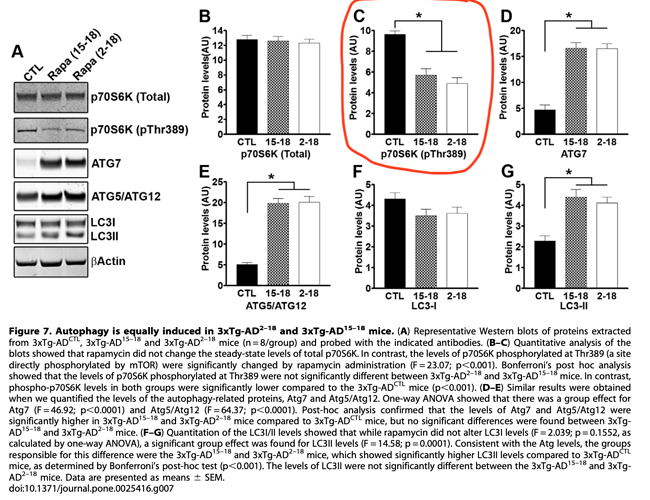
This study did NOT measure levels of Rapamycin in the brain. But they did measure pmTOR, showing significant decline in a direct measurement of mTOR inhibition.
Is this fundamentally therapeutic translatable, and how to even measure in humans?! Looks like 30-50% mTOR inhibition in the brain is a necessary signal. We talk about “turning off mTOR”, yet we might need “some constant level” mTOR reduction in the brain for therapeutic efficacy?? I think this “off logic” is misguided rationale.
“Overall, our data show that under the conditions used here, facilitating autophagy induction prophylactically has beneficial effects on AD-like pathology; however, once plaques and tangles are well established, increasing autophagy induction is not sufficient to rescue AD-like pathology and the associated cognitive deficits”
“The role of autophagy in AD is controversial. For example, Nixon and colleagues have shown that in AD brains there is accumulation of autophagosomes. Additionally, they showed that autophagosomes may be another source of Ab generation, suggesting that interventions aimed at further increasing autophagy induction in AD may actually exacerbate the Ab pathology. In contrast, Wyss-Coray and colleagues have shown that increasing autophagy induction decreases Ab pathology in an animal model of AD. Data in apparent contradiction to each other have been also reported by others. Our data are compatible with both views as we show that increasing autophagy induction prior to the development of AD like pathology in the 3xTg-AD mice reduces the levels of soluble Ab and tau and the formation of thioflavin-positive plaques. In contrast, we show that if autophagy is induced after mature plaques and tangles are formed, no changes in Ab, tau or cognitive deficits are detected. We suggest that increasing autophagy induction may be a valid therapeutic strategy for AD if the intervention occurs early in the development of the disease. In contrast, once the neuropathology is well-established, increasing autophagy induction alone may not be sufficient to ameliorate the neuropathological phenotype and different approaches should be considered. Toward this end, recently it has been shown that reversing autophagy dysfunction by increasing lysosomal function, is a good strategy in AD”
We still do NOT understand the true upstream etiology of AD, but clearly once aggregate plaques and tangles are formed (in humans, extending translation of this mouse model), these don’t appear reversible and disrupt cognitive signalling.
Regarding timing and efficacy of Rapamycin administration to slow cognitive decline, that’s a very difficult question to answer.
It would depend on amyloid and plaque load, and some “arbitrary” cutoff definition re incipient acceleration from normal cognitive to MCI. This would require full brain scans for glucose hypometabolism, amyloid and tau. This is NOT standard imaging (very $$), typically only done in a trial setting, high risk patients. These data “might” assist with snapshot and risk trajectory assessment and whether Rapamycin is a proper risk/reward. This would need experienced AD focused neurologist up on the very latest treatment options. And even so, still a guess.
From:
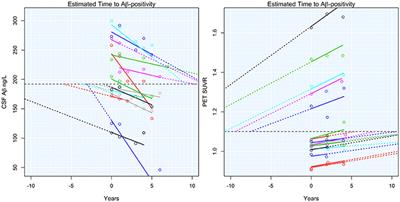
Time to Amyloid Positivity and Preclinical Changes in Brain Metabolism, Atrophy, and Cognition: Evidence for Emerging Amyloid Pathology in Alzheimer’s Disease
Cerebrospinal fluid (CSF) biomarkers and positron emission tomography (PET) with tracers sensitive to Aβ, have made it possible to study the development of AD-related brain changes in the brain of living humans. However, it is not clear at what degree of Aβ pathology downstream AD-related changes start to occur. For example, is it necessary to have fully established Aβ pathology before any changes in metabolism can be detected, or can such changes occur much earlier, before conventional thresholds for Aβ+ are reached? Taken together, the studies discussed above suggest that there may be an early stage of abnormal Aβ-metabolism, when biomarker signs of Aβ-pathology start to appear and are related to other aspects of AD, even before they become Aβ+, i.e., prior to meeting the conventional criteria for preclinical AD. We call this stage of disease “emerging Aβ-pathology.”
Decline in delayed memory recall was estimated to begin accelerating 10 years before Aβ+, with global measures of cognition following 5–8 years later
From:
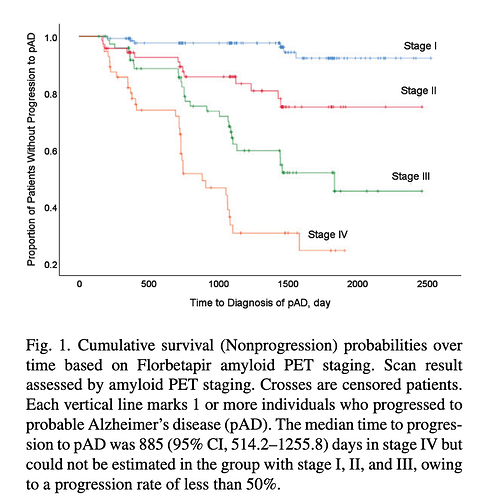
Quantitative Brain Amyloid Measures Predict Time-to-Progression from Amnestic Mild Cognitive Impairment to Alzheimer’s Disease
“To assess the prognostic impact of amyloid burden, a staging system based on the global SUVr of the PET scan was applied. We defined the stages as: stage I, negative amyloid scan; stage II, positive amyloid in 1st tertile; stage III, positive amyloid in 2nd tertile; and stage IV, positive amyloid in 3rd tertile.”
Conclusions: Large amyloid burden measured from amyloid PET scan could be a predictor of faster cognitive decline in aMCI patients.