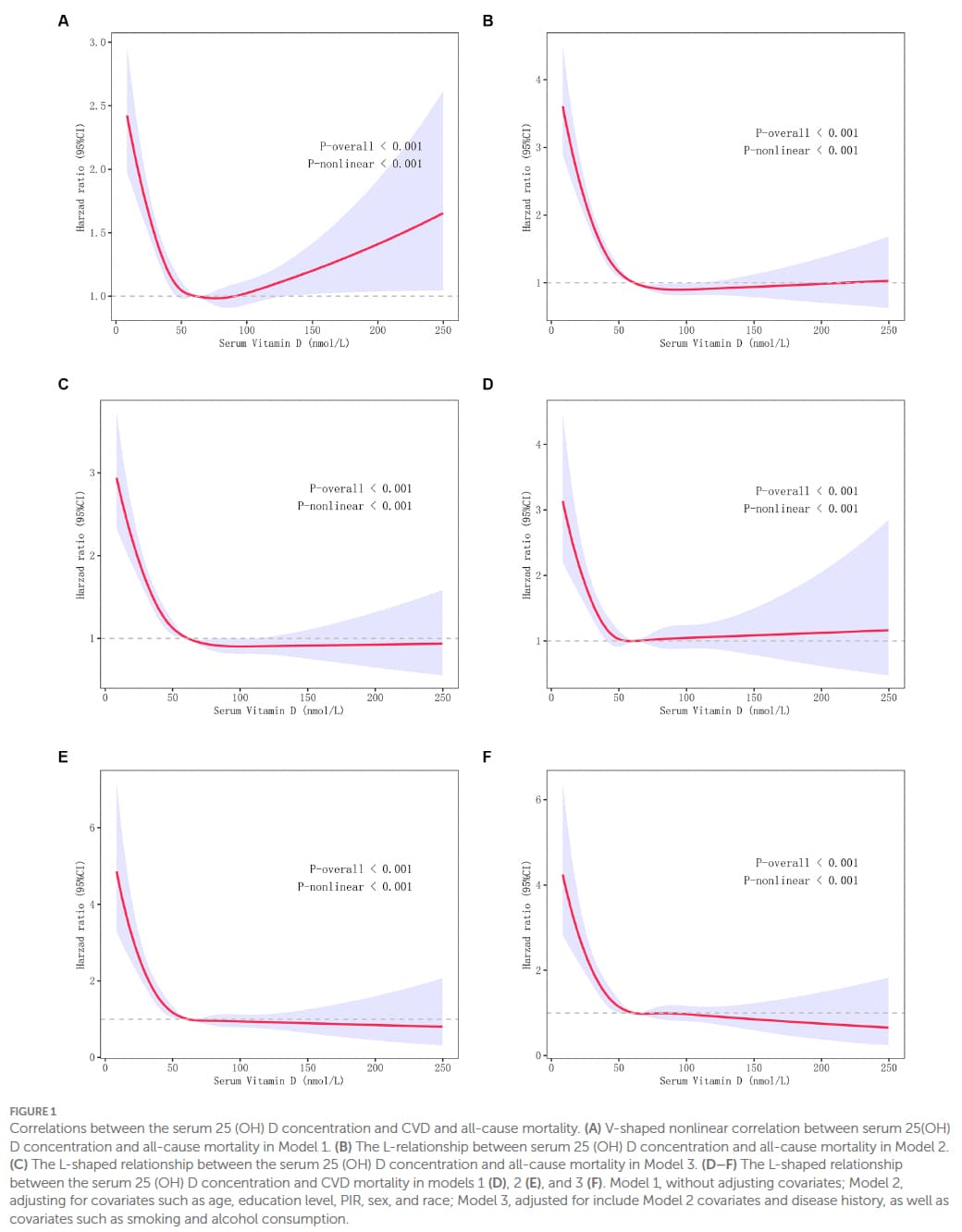
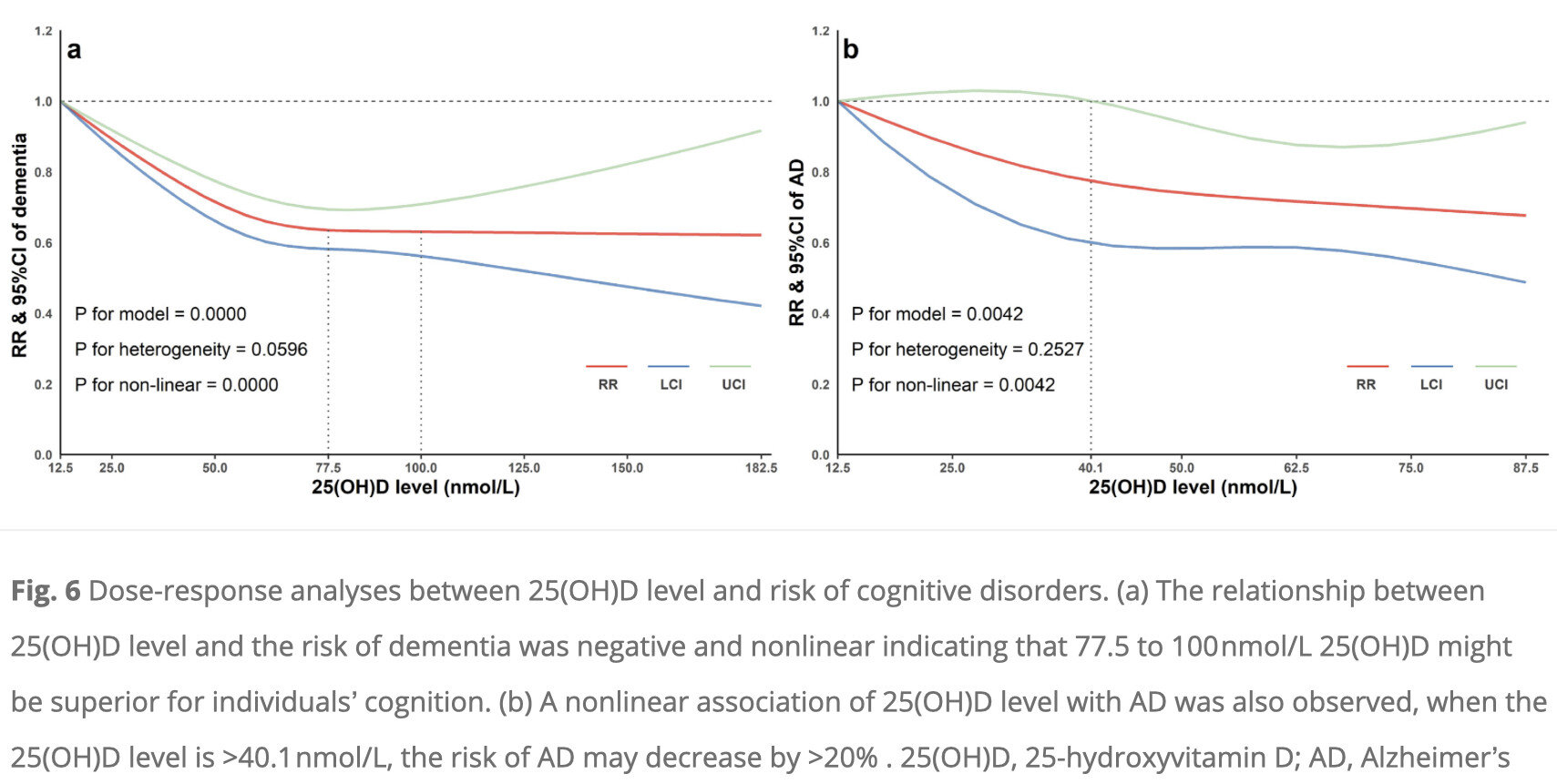
Looking at the paper:
According to the subgroup analysis of sex and age, the serum 25(OH)D concentration in the ≥60-year-old (p < 0.001 for trend) and female (p < 0.001 for trend) populations also showed an upward trend. Conversely, in the cohort aged 20–39 years, the exhibited a decreasing serum 25(OH)D concentration decreased trajectory from 66.2 nmol/L (95% CI, 64.3–68.1 nmol/L) in 2001–2002 to 61.9 nmol/L (95% CI, 59.1–64.8 nmol/L) in 2017–2018 (p < 0.002 for trend; Table 2).
I have had a good look through and it does not appear to adjust the chart reported by age. However, I have not spent a lot of time on this and may be wrong.
1 Like
cl-user
#584
That’s not at all what those papers show.
I don’t know why @adssx only showed the unadjusted model but it’s not at all what the better models which are adjusted for confounders show.
Here is the full picture. As you can see, higher serum vitamin D is better in all the adjusted models.
BTW the paper you cited also shows similar L shaped plots:
Basically too low vitamin D is universally bad while higher levels are neutral or trending better
3 Likes
I would say that it is clear that deficiency is bad. There is, however, uncertainty as to whether higher levels are better. If they are there is a marginal benefit.
I do wonder in this about cholecalciferol. There have been bolus monthly tests which have found no benefit and that will not have been adjusted for.
2 Likes
adssx
#587
Which paper? (20 char limit)
Yes and those later models show a different outcome.
I think we agree that vitamin D is essential up to say 75 nmol/L. After that point I think there is an advantage up to the higher levels even if a small advantage, but that people should avoid taking too much cholecalciferol as it is mildly toxic. You think people should stick at 75 nmol/L.
I would like to see charts adjusted for the metabolism issues related to cholecalciferol being converted into calcifediol, but I don’t think anyone is doing that.
Personally I may experiment for a few weeks with higher levels at some stage (once I get my next stash of calcifediol which is in the post). I may also experiment with dropping cholecalciferol supplementation and only supplementing with calcifediol.
I did some d3 experimentation in some detail about 3-4 years ago and I could feel when my body ran out of calcifediol a few days after stopping cholecalciferol supplementation.
One factor I like to consider is mechanism. As I see it the key mechanism for D3 is the Vitamin D Receptor and particularly its role as a transcription factor.
1 Like
adssx
#590
Actually, what I think is:
- Supplement if below 75 nmol/L (30 ng/mL)
- If above that, there might still be benefits in supplementing, but as of today, it’s unclear. In any case there are so many other things that are more important that people should focus their energy there and not on vitamin D (apoB, blood pressure, HbA1C, HRV, sleep, vitamin Bs, etc.). The time spent by people I know (friends and family) on “optimizing” vitamin D is insane compared to the unproven benefits (beyond deficiency, again). It’s a net negative when you consider the opportunity cost of not focusing on more important biomarkers. But if someone is perfect everywhere, then yes they might want to “optimize” vitamin D (which would first require to find the optimal serum levels AND the optimum form and dosage of supplementation).
1 Like
I don’t think we actually disagree on this. I still tend to aim for 200 nmol/L (80 ng/mL), but it is not my top priority.
I don’t think vitamin D lies on any of the key longevity pathways, but it does help the body function more effectively.
From an experimental basis I am interested in seeing if there are any identifiable effects of running at 300 or 400 nmol/L, but this is not a priority. I have done it unintentionally in the past.
1 Like
adssx
#592
Yes, I think we agree, I just wanted to make it clear to the silent majority of readers 
1 Like
Large-scale proteomic analyses of incident Parkinson’s disease reveal new pathophysiological insights and potential biomarkers.
https://www.nature.com/articles/s43587-025-00818-0
3 Likes
cl-user
#594
I was replying to @DrFraser who mentioned that paper below:
1 Like
adssx
#595
Thanks for sharing!
This study provides valuable insights into the dynamic changes of PD-associated proteins over time, bridging an important knowledge gap in the natural history of sporadic PD. We observed early and consistent lipid metabolism abnormalities during the pre-PD period, adding new evidence to the relationship between lipid metabolism-related proteins and PD. Previous studies noted alterations in lipid metabolism involving the carnitine shuttle, sphingolipid metabolism, arachidonic acid metabolism and fatty acid biosynthesis in patients with PD44. Lipid biology is strongly associated with α-synuclein physiology and pathology, central to PD pathogenesis. Our findings suggest that lipid metabolism abnormalities might start at least 15 years before disease onset, indicating a potential bidirectional relationship among lipid dyshomeostasis, abnormal α-synuclein accumulation and PD. We observed early functional abnormalities in B cell activation, neurosecretion, neurogenesis, immune response and ECM organization. Although individual protein changes were not sustained, these functional abnormalities persisted throughout PD. Our findings align with the hypothesis that inflammation and immune dysfunction, interacting with genetic and environmental factors, enable PD development and progression. Genetic and epidemiological evidence supports this hypothesis.
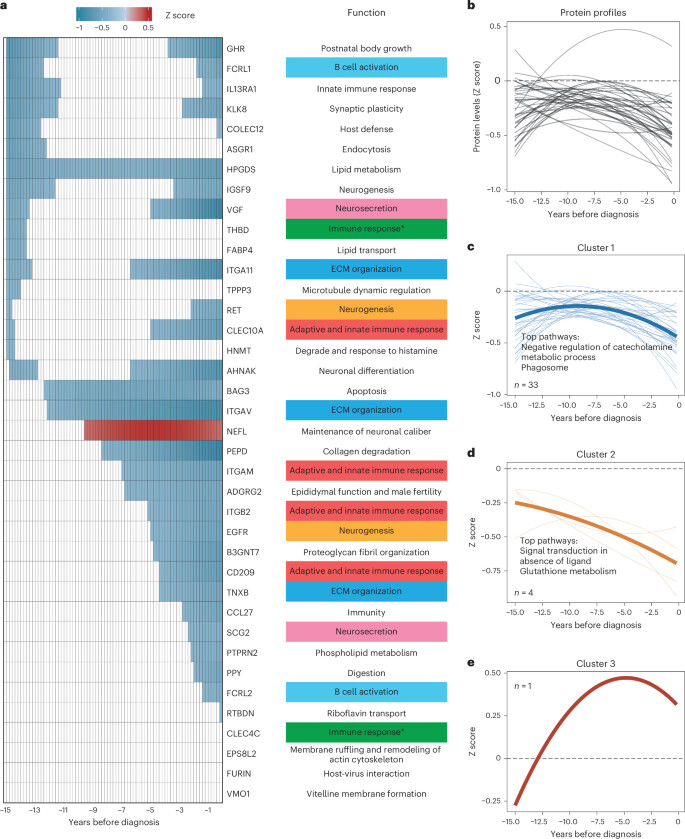
We observed that plasma HPGDS was the first protein to reach the abnormality 15 years before diagnosis and maintained an abnormally low level throughout the course (Fig. 3a). Based on the results of functional annotations for network categories (Fig. 2g) or the identification of nonclustered orphan proteins (Supplementary Table 15), we categorized each protein into its biological context. The function of the HPGDS protein was enriched in lipid metabolism, suggesting that abnormalities associated with lipid metabolism first appeared and might continue to intensify. The other 16 proteins also reached the defined abnormality about 15 years before diagnosis, but their change in levels did not persist throughout the course (Fig. 3a). The decreases in GHR, FCRL1, IL13RA1, KLKB, COLEC12, IGSF9, VGF, ITGA11, RET, CLEC10A and AHNAK sustained 0.2 to 3.6 years from −15 years, followed by recovery to normality, but their levels again decreased when approaching diagnosis. ASGR1, THBD, FABP4, TPPP3 and HNMT also experienced similar changes around −15 years, followed by similar Z score levels compared with controls. We noticed that BAG3 and ITGAV also reached an abnormally low level before −12 years. A significant portion of the remaining protein, including NEFL, PEPD, ITGAM, ADGRG2, ITGB2 and epidermal growth factor receptor (EGFR) had also reached the abnormality 5 years before diagnosis. The results of functional annotations for those 16 proteins (Fig. 3a) indicated that other anomalous activities, which may be transient in the early stages and exacerbated in the late stages, included B cell activation, neurosecretion, neurogenesis, immune response and ECM organization.
Our cis-MR findings suggested that genetic predisposition to upregulated EGFR was associated with a lower risk of PD (odds ratio (95% confidence interval (CI)) = 0.76 (0.63,0.91); P = 0.003), and higher genetically predicted level of ITGAM was associated with a higher risk of PD (odds ratio (95% CI) = 1.25 (1.01,1.55); P = 0.041). No significant relationship remained after false discovery rate correction (Supplementary Table 30).
ITGAM, also known as CD11B, is a key regulator of microglial activation and mediates locus coeruleus neurodegeneration via NLRP3 inflammasome-driven inflammation and oxidative stress in a PD mouse model. The relationships between these proteins and early symptoms strengthened the associations. Risk of RBD, a common sleep disorder in prodromal stage, was positively linked to elevated serum ITGAM level. Although limited, previous study shows that ITGAM assessment is associated with sleep deprivation
Our cis-MR findings suggested genetically predicted levels of EGFR and ITGAM were nominally associated with PD risk. The relationship between ITGAM and PD appeared contradictory in our Cox-regression and MR analyses, similar to previous studies. This paradox may arise from feedback mechanisms or other complex processes, requiring further investigation. Although EGFR’s causal relationship with PD did not remain significant after false discovery rate correction, its SNP’s previous association with PD susceptibility warrants further research on its potential as a target for PD pathogenesis and drug development.
So what can upregulate EGFR (epidermal growth factor receptor) and downregulate ITGAM (integrin alpha-M)? 
1 Like
Find yourself a lactating lady??
Epidermal Growth Factor
Epidermal growth factor (EGF) is a small polypeptide mitogen that has been identified in many species and isolated and characterized in human milk.
https://www.sciencedirect.com/topics/medicine-and-dentistry/epidermal-growth-factor
3 Likes
adssx
#597
Royal jelly / royalactin might be easier: Royalactin extends lifespan of Caenorhabditis elegans through epidermal growth factor signaling 2014
Whereas RJ is a complex mixture of sugars, proteins, lipids and vitamins (Takenaka, 1982), it was recently shown that the RJ protein royalactin is essential for queen differentiation (Kamakura, 2011). Royalactin activates the epidermal growth factor receptor (EGFR) signaling pathway in honeybee larvae, which ultimately leads to epigenetic changes and queen development. Moreover, Kamakura (2011) showed that royalactin shortens development time and increases longevity in the fruit fly Drosophila melanogaster through the EGFR signaling pathway, thus demonstrating that the glycoprotein also confers its beneficial effects on another insect species.
RJ has previously been shown to extend lifespan in mice (Inoue, 2003) and in the nematode Caenorhabditis elegans (Honda et al., 2011). However, the primary factor responsible for this lifespan extension remained elusive. Interestingly, one of the main conclusions drawn from the study on C. elegans was that RJ proteins are not likely contributors to the lifespan-extending effect of RJ (Honda et al., 2011), implying that royalactin may not be able to extend lifespan in C. elegans.
3 Likes
cl-user
#598
Ergothioneine is very interesting with a lot of new studies coming out like that one (Chinese, paywalled):
Ergothioneine exerts neuroprotective effects in Parkinson’s disease: Targeting α-synuclein aggregation and oxidative stress
Ergothioneine (EGT) is a natural dietary antioxidant derived from certain edible mushrooms, commonly used as a food additive and supplement, but its effects on Parkinson’s Disease (PD) are still unclear. The accumulation of α-synuclein (α-syn) plays a pivotal role in the pathogenesis and development of PD. Here, this study demonstrated that EGT effectively inhibits α-syn aggregation, disrupts mature fibers, and reduces associated cytotoxicity and oxidative stress. The beneficial effects of EGT were confirmed in Caenorhabditis elegans , where it protected dopaminergic neurons, prolonged lifespan and enhanced behavioral functions by reducing α-syn plaque accumulation and associated oxidative stress. Molecular dynamics simulation revealed that EGT interacts directly with α-syn pentamer through van der Waals and electrostatic forces, disrupting the structural stability of the preformed pentamer. Furthermore, animal studies validated that EGT alleviated neuronal damage and improved behavioral deficits by reducing α-syn aggregation, oxidative stress and inflammatory response. In conclusion, EGT presents promising potential as a dietary supplement for preventing and alleviating PD.
Another one: Ergothioneine-Mediated Neuroprotection of Human iPSC-Derived Dopaminergic Neurons
Cell death involving oxidative stress and mitochondrial dysfunction is a major cause of dopaminergic neuronal loss in the substantia nigra (SN) of Parkinson’s disease patients. Ergothioneine (ET), a natural dietary compound, has been shown to have cytoprotective functions, but neuroprotective actions against PD have not been well established.[…]
These results suggest that ET could be a potential therapeutic for Parkinson’s disease.
2 Likes
adssx
#599
Thanks. I need to do more research on that but the issue is that both papers looked at toxin models (MPTP and 6-OHDA). PD has been cured hundreds of times on these models. They don’t represent the disease well. Transgenic models are better (the first paper mentions a transgenic model of C Elegans but doesn’t say which strain). So when it works on a toxin model it’s an interesting signal but nothing more.
I did a quick search on EGT and found: Food-derived antioxidant ergothioneine improves sleep difficulties in humans 2022
“In addition, EGT showed inhibitory activity of ≥ 25% and ≤ 50% at a concentration of ≤ 1,000 μM against seven molecular targets [epidermal growth factor receptor”
If I’m correct we want to upregulate EGFR, not inhibit it. So if this paper is also correct then EGT is highly detrimental in PD. No?
4 Likes
cl-user
#600
Interesting paper. I view it as a generally positive for EGT as it improves sleep even though this might not be useful for PD.
Concerning the EGFR inhibition it’s 43% at 1000µM and 10% at 100µM but these are very high concentration tested in vitro. The levels they got in humans with a 20mg/day supplementation are much lower at 9.54µM.
Obviously finding the optimal levels of interventions for a complex disease like PD is a pareto optimization problem (i.e. the global optimal solution is a tradeoff and not the combination of the optimal for each intervention) and there is no easy way to find it.
1 Like
9-Methyl-β-carboline inhibits monoamine oxidase activity and stimulates the expression of neurotrophic factors by astrocytes
https://www.researchgate.net/publication/5932301_9-Methyl-beta-carboline_up-regulates_the_appearance_of_differentiated_dopaminergic_neurones_in_primary_mesencephalic_culture
1 Like