I personally find the relocalization of chromatin modifiers (RCM) hypothesis is one of the most convincing mechanisms of epigenetic aging (and also aging in general), and there’s evidence that DSBs are involved, in a manner that seems mostly independent of their ability to produce insertions and deletions.
The exact nature of DSB-induced changes in chromatin structure and how these structural changes might impart age-associated transcriptional changes (accelerated elongation and increased stalling of RNA polymerase) isn’t clear, but it’s plausible that changes in chromatin structure could produce such a transcriptional phenotype, or partially contribute to it.
Despite this, the primary contributor to age-associated RNA polymerase stalling appears to be DNA damage, perhaps of an oxidative variety.
Subsequently, we assessed whether various potential parameters were correlated with the degree of GLPT to identify a mechanism explaining RNAPII stalling. We did not find significant differences between GLPThigh genes and other gene categories (transcriptionally upregulated and downregulated; remainder) in nucleotide content across gene bodies, transcriptional error rate, alternative splicing, chromatin accessibility, histone modifications associated with euchromatin or DNA methylation patterns, which would point to epigenetic changes being responsible (Extended Data Figs. 4–6). These factors do not correlate with the degree of age-related GLPT, which is expected when such a factor is causally involved, and hence do not explain the observed transcriptional decline.
In view of gene-length-dependent transcriptional stalling, a plausible explanation is accumulation of transcription-blocking DNA damage because long genes have a higher probability to acquire stochastic lesions26,29. Therefore, we monitored de novo RNA synthesis in the livers of Xpg −/− mice, which display many features of widespread premature aging and a 20-week life span due to defects in the DNA repair pathways TCR and global genome nucleotide excision repair by which they are unable to remove transcription-stalling lesions34. Both EU staining and EU-seq at the age of 7 and 14 weeks (Fig. 4a–c) revealed an age-dependent, progressive, pan-nuclear decline in transcription. EU-seq in premature aging global genome nucleotide excision repair and TCR-defective Ercc1 Δ/− mice that display more severe liver aging pathology due to an additional defect in interstrand cross-link repair35,36, exhibited already high transcription loss at 4 and even more at 10 weeks (Fig. 4d), showing that the extent of transcriptional decline, DNA repair deficiency and severity of liver pathology are correlated. [ref]
As stated above, Xpg is a component of the nucleotide excision repair pathway, and is known to remove oxidative lesions like 5-guanidinohydantoin and spiroiminodihydantoin (both of which form from 8-oxoguanine), and both of which have been shown to stall RNA polymerase II.
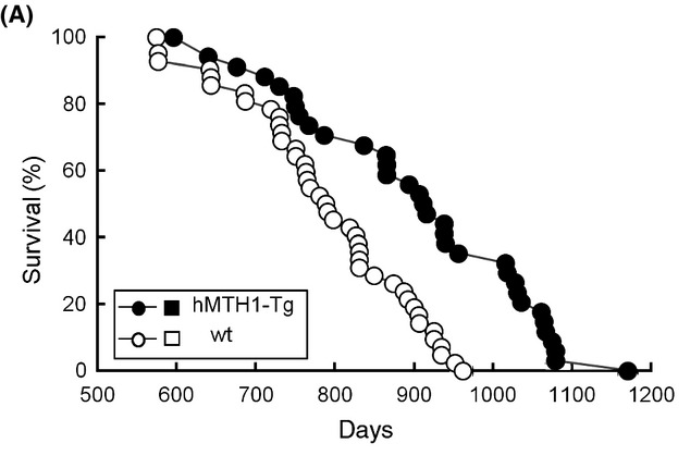
This is supported by increased lifespan in hMTH1 transgenic mice, although ideally this would be shown in genetically heterogenous mice with control median lifespan >850 days
hMTH1-Tg mice express high levels of the hMTH1 hydrolase that degrades 8-oxodGTP and 8-oxoGTP and excludes 8-oxoguanine from both DNA and RNA.
…
hMTH1-Tg animals live longer. Their median lifespan was 4 months greater than that of their wild-type littermates (914 vs. 790 days) (P < 0.0004; Kaplan Meier test) (Fig. 1A). [ref]
Also interesting to note that the age-associated changes in elongation rate appear to arise from histone and nucleosome depletion. As DNA G-quadruplexes have been shown to increase with replicative aging and form on nucleosome-free DNA, I wonder if they might be contribute to RNAPII stalling.
There’s not a ton of research on this and G-quadruplexes close to the transcription start site appear to stimulate transcription. OTOH, they can also produce an absolute transcriptional block, and I wonder if G-quadruplexes further downstream in the gene body might produce stalling. Presumably the average number of G-quadruplexes in the gene body would be proportional to its length, so this could explain the length-dependence of transcriptional changes.