Chris, @J0hn and others thinking about the differences I looked a bit further into that and the more I look at it the more it does seem like one might want to SGLT1 also is not the same as SGLT2*.
—
On the fact that SGLT1 is not the same as SGLT2* - not only that are they very different mechanistically, but also outcome wise have differences:
Thus far, the results from SCORED and SOLOIST (trials studying the SGLT1/2 inhibitor sotagliflozin) suggest that an increase in SGLT1 inhibition when added to SGLT2 inhibition may contribute to reductions in MI and stroke in patients with T2DM. This benefit is beyond what SGLT2is alone can accomplish
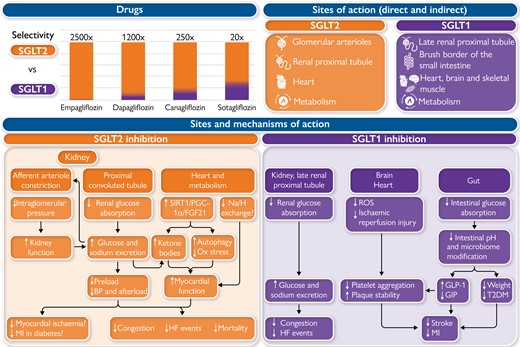
There are, however, major differences in the relative inhibition of SGLT1 and SGLT2 among the SGLTis.
Sodium glucose cotransporter-2 is a high capacity, low-affinity glucose transporter expressed in the proximal renal tubule where it is responsible for ∼97% of urinary glucose absorption and 4–5% of urinary sodium absorption in normal individuals, and its inhibition results in an increase in glucose and sodium excretion. Sodium glucose cotransporter-2 inhibition has been shown to be effective in preventing as well as treating HF, both HFrEF and HFpEF, in high CV risk patients with as well as in those without T2DM, and also in patients with HF but without T2DM.1 They may exert their beneficial effects through multiple mechanisms in the kidneys and myocardium and via altered metabolism. At the kidney level, SGLT2 inhibitors cause osmotic diuresis and natriuresis without renin–angiotensin system and sympathetic nervous system activation and with restored tubuloglomerular feedback, glomerular afferent arteriole constriction, decreased intraglomerular perfusion pressure, and increased erythropoietin secretion. Causing glycosuria, they lead to mild ketogenesis which increases myocardial efficiency. In addition, they inhibit sodium-hydrogen exchange, activate sirtuins, and induce autophagy in the cardiomyocytes All these mechanisms are potentially favourable for slowing the progression of chronic kidney disease (CKD) and HF. However, it is unclear which of them has a leading role. It remains true that SGLT2is are relatively well tolerated and have the potential to have a major impact on CV and renal outcomes in patients with HF +/− T2DM and +/− CKD.
Sodium glucose cotransporter-2 inhibition has, however, some limitations. For example, SGLT2is have not been associated with a reduction in non-fatal and fatal stroke in patients with T2DM despite a significant, although moderate reduction of about 3 mmHg in systolic blood pressure which, on the basis of prior epidemiologic observations and randomized trials in patients with hypertension, should have resulted in a reduction in stroke.4 A recent meta-analysis has shown a reduction in systolic blood pressure with SGLT2is in patients with diabetes by a weighted mean difference and 95% confidence intervals (CIs) of −2.89 (−3.37 to −2.40) mmHg.5 However, this was not associated with a change in the rate of stroke [relative risk (RR), 0.98, 95% CI, 86–1.11; P = 0.72] with a modest 10% reduction in the rate of myocardial infarction (RR, 0.90, 95% CI, 82–0.99; P = 0.03) in other meta-analyses.4,6,7 The reduction in MI associated with SGLT2i may be secondary to a reduction in preload and myocardial oxygen demands, which, in a patient with ischaemic heart disease, could reduce the risk of ischaemia and MI. Thus, both the lack of reduction in the rate of stroke and the modest reduction in MI may suggest lack of a primary anti-atherosclerotic or anti-thrombotic effect of SGLT2is.
Sodium glucose cotransporter-1 is a low capacity, high-affinity glucose transporter expressed in the late renal proximal tubule, where it is responsible for absorbing around 3% of urinary glucose in normal individuals. It is also expressed in the capillaries of the heart, brain, and skeletal muscle and, mainly, in the brush border of the small intestine where its inhibition results in increased delivery of glucose to the distal intestines with a decrease in intestinal pH and, as a result, alteration in the intestinal microbiome with an increase in short chain fatty acids and an increase in the secretion of the incretin glucagon-like peptide-1 (GLP-1) and a decrease in glucose-dependent insulinotropic polypeptide (GIP).8 The potential mechanisms by which SGLT1i reduce stroke and MI have been previously reviewed and include the finding that an increase in GLP-1 results in a decrease in platelet activation and an increase in atherosclerotic plaque stability.8,9 In preclinical models, SGLT1 has also been found in several other organs. However, despite these differences, controversy regarding the specificity of the antibodies used to detect SGLT1, the lack of a clear understanding of the function of SGLT1 in these tissues, and the likelihood that SGLT1 expression can increase in various tissues depending upon comorbidities such as HF and T2DM, suggest the need for further investigation of SGLT1i and SGLT1/2i.
The SGLT1/2i sotagliflozin increases urinary glucose persistently and urinary sodium excretion transiently and affects several other mechanisms similar to the SGLT2is in patients with normal renal function as well as in those with moderate renal dysfunction. However, since SGLT1 is also expressed in the intestines, its inhibition can delay glucose absorption even in patients with severe kidney disease. While a reduction in serum glucose does not appear to be critical in the prevention or treatment of HF in T2DM, it is likely of importance in the long-term prevention of the microvascular effects of T2DM such as retinopathy and neuropathy. The effects of SGLT1 inhibition on the microbiome in patients with T2DM and HF are incompletely investigated, but could also be of importance since alteration of the microbiome is associated with hypertension and the risk of stroke.9–11 Importantly, SGLT1 has been shown to be upregulated in patients with diabetic cardiomyopathy12 and is associated with an increase in NADPH oxidase 2 and, as a consequence, an increase in reactive oxygen species in models of myocardial ischaemia, whereas SGLT1 knock down attenuates ischaemic/reperfusion injury.13 Mendelian randomization studies examining missense variants associated with a decrease in SGLT1 function have been associated with a decrease in the incidence of HF.14 This may be related in part to the effect of SGLT1i on the release of GLP1 and GIP, both of which have an effect on appetite and weight loss, and could be of importance for the prevention of HF in view of the finding that individuals with T2DM and abdominal obesity have an increased risk of developing HF compared with those with T2DM without abdominal obesity and those with abdominal obesity but without T2DM.14
Does anyone have a sense for how big the GLP-1 effect is from SGLT1i? I’d not want to have a constant increase in insulin…. Like one can get from taking ozempic, etc